fitgmdist
가우스 혼합 모델을 데이터에 피팅
설명
예제
두 개의 혼합된 이변량 가우스 분포에서 데이터를 생성합니다.
mu1 = [1 2];
Sigma1 = [2 0; 0 0.5];
mu2 = [-3 -5];
Sigma2 = [1 0;0 1];
rng(1); % For reproducibility
X = [mvnrnd(mu1,Sigma1,1000); mvnrnd(mu2,Sigma2,1000)];가우스 혼합 모델을 피팅합니다. 두 개의 성분이 있다고 지정합니다.
GMModel = fitgmdist(X,2);
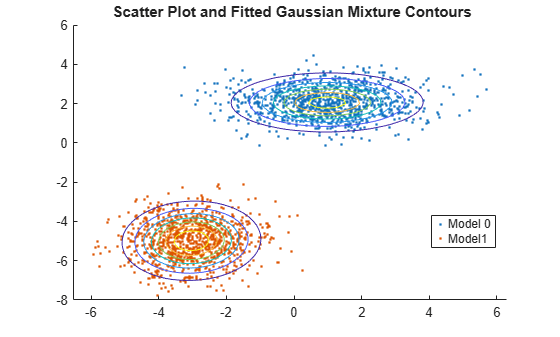
피팅된 가우스 혼합 모델 등고선에 데이터를 플로팅합니다.
figure y = [zeros(1000,1);ones(1000,1)]; h = gscatter(X(:,1),X(:,2),y); hold on gmPDF = @(x,y) arrayfun(@(x0,y0) pdf(GMModel,[x0 y0]),x,y); g = gca; fcontour(gmPDF,[g.XLim g.YLim]) title('{\bf Scatter Plot and Fitted Gaussian Mixture Contours}') legend(h,'Model 0','Model1') hold off
두 개의 혼합된 이변량 가우스 분포에서 데이터를 생성합니다. 첫 번째 예측 변수와 두 번째 예측 변수의 합인 세 번째 예측 변수를 생성합니다.
mu1 = [1 2];
Sigma1 = [1 0; 0 1];
mu2 = [3 4];
Sigma2 = [0.5 0; 0 0.5];
rng(3); % For reproducibility
X1 = [mvnrnd(mu1,Sigma1,100);mvnrnd(mu2,Sigma2,100)];
X = [X1,X1(:,1)+X1(:,2)];X의 열은 선형 종속입니다. 이는 조건이 나쁜 공분산 추정값을 초래할 수 있습니다.
가우스 혼합 모델을 데이터에 피팅합니다. try/catch 문을 사용하면 오류 메시지를 관리하는 데 도움이 될 수 있습니다.
rng(1); % Reset seed for common start values try GMModel = fitgmdist(X,2) catch exception disp('There was an error fitting the Gaussian mixture model') error = exception.message end
There was an error fitting the Gaussian mixture model
error = 'Ill-conditioned covariance created at iteration 2.'
공분산 추정값은 조건이 나쁩니다. 따라서, 최적화가 중지되고 오류가 표시됩니다.
가우스 혼합 모델을 다시 피팅하되, 이번에는 정규화를 사용합니다.
rng(3); % Reset seed for common start values GMModel = fitgmdist(X,2,'RegularizationValue',0.1)
GMModel = Gaussian mixture distribution with 2 components in 3 dimensions Component 1: Mixing proportion: 0.536725 Mean: 2.8831 3.9506 6.8338 Component 2: Mixing proportion: 0.463275 Mean: 0.8813 1.9758 2.8571
이 경우, 알고리즘이 정규화로 인해 해로 수렴합니다.
가우스 혼합 모델을 사용하려면 데이터에 피팅하기 전에 성분의 개수를 지정해야 합니다. 많은 응용 사례의 경우 적합한 성분 개수를 파악하는 것이 어려울 수 있습니다. 이 예제에서는 주성분 분석을 사용하여 데이터를 탐색하고 성분의 개수에 대한 초기 추측값을 구하는 방법을 보여줍니다.
피셔(Fisher)의 붓꽃 데이터 세트를 불러옵니다.
load fisheriris
classes = unique(species)classes = 3×1 cell
{'setosa' }
{'versicolor'}
{'virginica' }
데이터 세트에는 붓꽃 종에 대한 세 개 클래스가 있습니다. 분석은 마치 이런 사실을 모르는 것처럼 진행됩니다.
시각화를 위해 데이터 차원을 2차원으로 줄이려면 주성분 분석을 사용하십시오.
[~,score] = pca(meas,'NumComponents',2);각각 1개, 2개, 3개 성분을 지정하여 세 개의 가우스 혼합 모델을 데이터에 피팅합니다. 최적화 반복 횟수를 1000으로 늘립니다. 점 표기법을 사용하여 최종 모수 추정값을 저장합니다. 기본적으로, 소프트웨어는 각 성분에 대해 완전 공분산과 여러 다른 공분산을 피팅합니다.
GMModels = cell(3,1); % Preallocation options = statset('MaxIter',1000); rng(1); % For reproducibility for j = 1:3 GMModels{j} = fitgmdist(score,j,'Options',options); fprintf('\n GM Mean for %i Component(s)\n',j) Mu = GMModels{j}.mu end
GM Mean for 1 Component(s)
Mu = 1×2
10-15 ×
0.9607 -0.4371
GM Mean for 2 Component(s)
Mu = 2×2
1.3212 -0.0954
-2.6424 0.1909
GM Mean for 3 Component(s)
Mu = 3×2
0.4856 -0.1287
1.4484 -0.0904
-2.6424 0.1909
GMModels는 세 가지 피팅된 gmdistribution 모델을 포함하는 셀형 배열입니다. 세 가지 성분 모델의 평균이 서로 다릅니다. 이는 모델이 세 가지 붓꽃 종을 구분한다는 것을 나타냅니다.
피팅된 가우스 혼합 모델 등고선에 점수를 플로팅합니다. 데이터 세트가 레이블을 포함하므로 성분의 실제 개수를 구분할 수 있도록 gscatter를 사용하십시오.
figure for j = 1:3 subplot(2,2,j) h1 = gscatter(score(:,1),score(:,2),species); h = gca; hold on gmPDF = @(x,y) arrayfun(@(x0,y0) pdf(GMModels{j},[x0 y0]),x,y); fcontour(gmPDF,[h.XLim h.YLim],'MeshDensity',100) title(sprintf('GM Model - %i Component(s)',j)); xlabel('1st principal component'); ylabel('2nd principal component'); if(j ~= 3) legend off; end hold off end g = legend(h1); g.Position = [0.7 0.25 0.1 0.1];
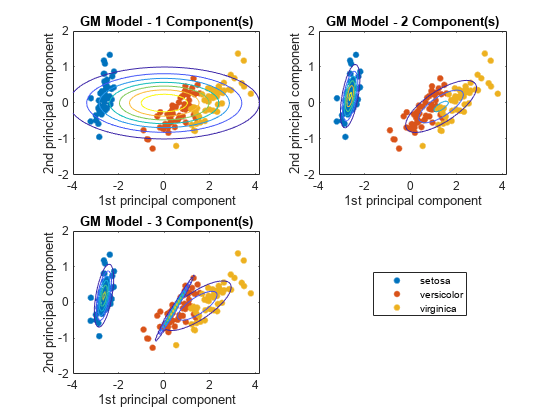
세 개 성분을 갖는 가우스 혼합 모델은 PCA와 함께 사용할 경우 세 가지 붓꽃 종을 구분하는 것처럼 보입니다.
가우스 혼합 모델에 적합한 성분 개수를 선택하는 데 도움이 되는 다른 옵션을 사용할 수 있습니다. 예를 들면 다음과 같습니다.
정보 기준(예: AIC 또는 BIC)을 사용하여 다양한 성분 개수를 갖는 여러 모델을 비교합니다.
Calinski-Harabasz 기준과 갭 통계량 또는 기타 기준을 지원하는
evalclusters를 사용하여 군집 개수를 추정합니다.
가우스 혼합 모델을 사용하려면 데이터에 피팅하기 전에 성분의 개수를 지정해야 합니다. 많은 응용 사례의 경우 적합한 성분 개수를 파악하는 것이 어려울 수 있습니다. 이 예제에서는 다양한 개수의 성분에서 최적의 피팅 가우스 혼합 모델을 선택하는 데 도움이 되도록 AIC 피팅 통계량을 사용합니다.
두 개의 혼합된 이변량 가우스 분포에서 데이터를 생성합니다.
mu1 = [1 1]; Sigma1 = [0.5 0; 0 0.5]; mu2 = [2 4]; Sigma2 = [0.2 0; 0 0.2]; rng(1); X = [mvnrnd(mu1,Sigma1,1000);mvnrnd(mu2,Sigma2,1000)]; plot(X(:,1),X(:,2),'ko') title('Scatter Plot') xlim([min(X(:)) max(X(:))]) % Make axes have the same scale ylim([min(X(:)) max(X(:))])
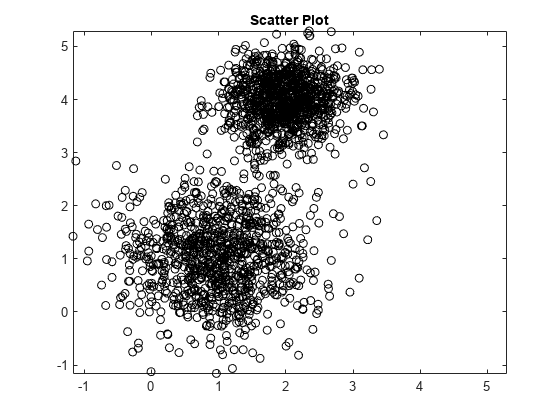
기본 모수 값을 알지 못한다고 가정해도 산점도 플롯을 보면 다음을 알 수 있습니다.
두 개 성분이 있습니다.
군집 간 분산이 다릅니다.
군집 내 분산이 같습니다.
군집 내에 공분산이 없습니다.
두 개 성분을 갖는 가우스 혼합 모델을 피팅합니다. 산점도 플롯을 관찰한 결과를 기반으로 하여 공분산 행렬이 대각 행렬임을 지정하십시오. statset 구조체를 Options 이름-값 쌍의 인수 값으로 전달하여 최종 반복 및 로그 가능도 통계량을 명령 창에 출력합니다.
options = statset('Display','final'); GMModel = fitgmdist(X,2,'CovarianceType','diagonal','Options',options);
11 iterations, log-likelihood = -4787.38
GMModel은 피팅된 gmdistribution 모델입니다.
다양한 개수의 성분에 대해 AIC를 검토합니다.
AIC = zeros(1,4); GMModels = cell(1,4); options = statset('MaxIter',500); for k = 1:4 GMModels{k} = fitgmdist(X,k,'Options',options,'CovarianceType','diagonal'); AIC(k)= GMModels{k}.AIC; end [minAIC,numComponents] = min(AIC); numComponents
numComponents = 2
BestModel = GMModels{numComponents}BestModel = Gaussian mixture distribution with 2 components in 2 dimensions Component 1: Mixing proportion: 0.501719 Mean: 1.9824 4.0013 Component 2: Mixing proportion: 0.498281 Mean: 0.9880 1.0511
두 개 성분을 갖는 가우스 혼합 모델을 피팅할 때 가장 작은 AIC가 발생합니다.
가우스 혼합 모델 모수 추정값은 초기값에 따라 달라질 수 있습니다. 이 예제에서는 fitgmdist를 사용하여 가우스 혼합 모델을 피팅할 때 초기값을 제어하는 방법을 보여줍니다.
피셔(Fisher)의 붓꽃 데이터 세트를 불러옵니다. 꽃잎 길이와 너비를 예측 변수로 사용합니다.
load fisheriris
X = meas(:,3:4);디폴트 초기값을 사용하여 가우스 혼합 모델을 데이터에 피팅합니다. 세 가지 벚꽃 종이 있으므로 k = 3으로 성분 개수를 지정합니다.
rng(10); % For reproducibility
GMModel1 = fitgmdist(X,3);기본적으로 소프트웨어는 다음을 수행합니다.
초기화를 위한 k-평균++ 알고리즘을 구현하여 k = 3인 초기 군집 중심을 선택합니다.
초기 공분산 행렬을 요소 (
j,j)가X(:,j)의 분산인 대각 행렬로 설정합니다.초기 혼합 비율을 균일하게 처리합니다.
각 관측값을 해당 레이블에 연결하여 가우스 혼합 모델을 피팅합니다.
y = ones(size(X,1),1); y(strcmp(species,'setosa')) = 2; y(strcmp(species,'virginica')) = 3; GMModel2 = fitgmdist(X,3,'Start',y);
초기 평균, 공분산 행렬 및 혼합 비율을 명시적으로 지정하여 가우스 혼합 모델을 피팅합니다.
Mu = [1 1; 2 2; 3 3]; Sigma(:,:,1) = [1 1; 1 2]; Sigma(:,:,2) = 2*[1 1; 1 2]; Sigma(:,:,3) = 3*[1 1; 1 2]; PComponents = [1/2,1/4,1/4]; S = struct('mu',Mu,'Sigma',Sigma,'ComponentProportion',PComponents); GMModel3 = fitgmdist(X,3,'Start',S);
gscatter를 사용하여 붓꽃 종 간을 구분하는 산점도 도식을 플로팅합니다. 각 모델에 대해 피팅된 가우스 혼합 모델 등고선을 플로팅합니다.
figure subplot(2,2,1) h = gscatter(X(:,1),X(:,2),species,[],'o',4); haxis = gca; xlim = haxis.XLim; ylim = haxis.YLim; d = (max([xlim ylim])-min([xlim ylim]))/1000; [X1Grid,X2Grid] = meshgrid(xlim(1):d:xlim(2),ylim(1):d:ylim(2)); hold on contour(X1Grid,X2Grid,reshape(pdf(GMModel1,[X1Grid(:) X2Grid(:)]),... size(X1Grid,1),size(X1Grid,2)),20) uistack(h,'top') title('{\bf Random Initial Values}'); xlabel('Sepal length'); ylabel('Sepal width'); legend off; hold off subplot(2,2,2) h = gscatter(X(:,1),X(:,2),species,[],'o',4); hold on contour(X1Grid,X2Grid,reshape(pdf(GMModel2,[X1Grid(:) X2Grid(:)]),... size(X1Grid,1),size(X1Grid,2)),20) uistack(h,'top') title('{\bf Initial Values from Labels}'); xlabel('Sepal length'); ylabel('Sepal width'); legend off hold off subplot(2,2,3) h = gscatter(X(:,1),X(:,2),species,[],'o',4); hold on contour(X1Grid,X2Grid,reshape(pdf(GMModel3,[X1Grid(:) X2Grid(:)]),... size(X1Grid,1),size(X1Grid,2)),20) uistack(h,'top') title('{\bf Initial Values from the Structure}'); xlabel('Sepal length'); ylabel('Sepal width'); legend('Location',[0.7,0.25,0.1,0.1]); hold off
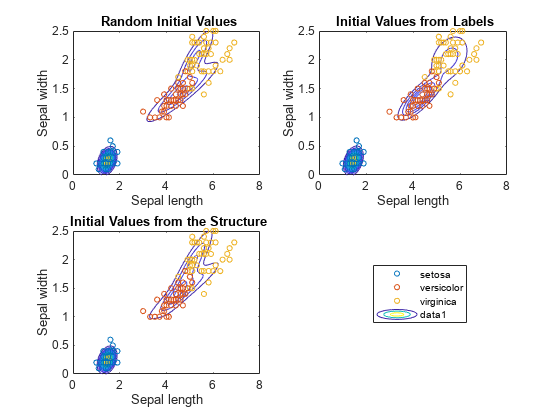
등고선을 보면 GMModel2는 약간의 삼봉 분포를 나타내는 것처럼 보이고, 나머지 모델은 이봉 분포를 나타냅니다.
추정된 성분 평균을 표시합니다.
table(GMModel1.mu,GMModel2.mu,GMModel3.mu,'VariableNames',... {'Model1','Model2','Model3'})
ans=3×3 table
Model1 Model2 Model3
_________________ ________________ ________________
5.2115 2.0119 4.2857 1.3339 1.4604 0.2429
1.461 0.24423 1.462 0.246 4.7509 1.4629
4.6829 1.4429 5.5507 2.0316 5.0158 1.8592
GMModel2가 붓꽃 종 간을 가장 잘 구분하는 것처럼 보입니다.
입력 인수
이름-값 인수
출력 인수
팁
fitgmdist 함수는 다음과 같을 수 있습니다.
하나 이상의 성분에 조건이 나쁜 공분산 행렬 또는 특이 공분산 행렬이 있는 경우 해로 수렴할 수 있습니다.
다음과 같은 문제로 인해 조건이 나쁜 공분산 행렬이 생성될 수 있습니다.
데이터의 차원 수가 비교적 높고 관측값 개수가 충분하지 않습니다.
데이터의 일부 예측 변수 간에 높은 상관관계가 있습니다.
일부 또는 모든 특징이 이산적입니다.
너무 많은 성분에 데이터를 피팅하려고 했습니다.
일반적으로 다음 예방책 중 하나를 사용하여 조건이 나쁜 공분산 행렬이 생성되는 것을 피할 수 있습니다.
데이터를 전처리하여 상관관계가 있는 특징을 제거합니다.
'SharedCovariance'를true로 설정하여 모든 성분에 동일한 공분산 행렬을 사용합니다.'CovarianceType'를'diagonal'로 설정합니다.'RegularizationValue'를 사용하여 매우 작은 양수를 모든 공분산 행렬의 대각선에 더합니다.다른 초기값 세트를 사용해 봅니다.
하나 이상의 성분에 조건이 나쁜 공분산 행렬이 있는 경우 중간 과정을 건너뛸 수 있습니다. 다른 초기값 세트를 사용하여 데이터 또는 모델을 변경하지 않고 이 문제를 방지할 수 있습니다.
알고리즘
참고 문헌
[1] McLachlan, G., and D. Peel. Finite Mixture Models. Hoboken, NJ: John Wiley & Sons, Inc., 2000.
버전 내역
R2014a에 개발됨