Exploring a Nucleotide Sequence Using Command Line
Overview of Example
After sequencing a piece of DNA, one of the first tasks is to investigate the nucleotide content in the sequence. Starting with a DNA sequence, this example uses sequence statistics functions to determine mono-, di-, and trinucleotide content, and to locate open reading frames.
Searching the Web for Sequence Information
The following procedure illustrates how to use the system web browser to search the Web for information. In this example you are interested in studying the human mitochondrial genome. While many genes that code for mitochondrial proteins are found in the cell nucleus, the mitochondrial has genes that code for proteins used to produce energy.
First research information about the human mitochondria and find the nucleotide sequence for the genome. Next, look at the nucleotide content for the entire sequence. And finally, determine open reading frames and extract specific gene sequences.
In the MATLAB® Command Window, type
web('http://www.ncbi.nlm.nih.gov/')A separate browser window opens with the home page for the NCBI Web site.
Search the NCBI Web site for information. For example, to search for the human mitochondrion genome, from the Search list, select
Genome, and in the Search list, entermitochondrion homo sapiens.
The NCBI Web search returns a list of links to relevant pages.
Select a result page. For example, click the link labeled NC_012920.
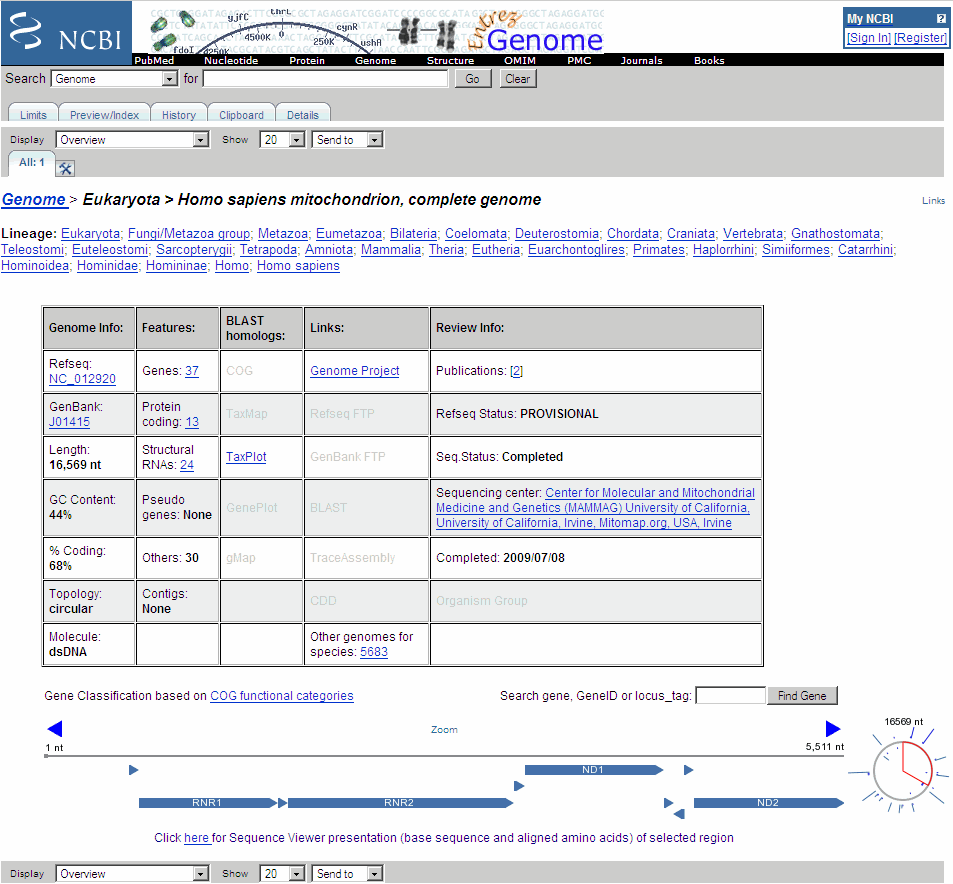
The web browser displays the NCBI page for the human mitochondrial genome.
Reading Sequence Information from the Web
The following procedure illustrates how to find a nucleotide sequence in a public database and read the sequence information into the MATLAB environment. Many public databases for nucleotide sequences are accessible from the Web. The MATLAB Command Window provides an integrated environment for bringing sequence information into the MATLAB environment.
The consensus sequence for the human mitochondrial genome has
the GenBank® accession number NC_012920. Since
the whole GenBank entry is quite large and you might only be
interested in the sequence, you can get just the sequence information.
Get sequence information from a Web database. For example, to retrieve sequence information for the human mitochondrial genome, in the MATLAB Command Window, type
mitochondria = getgenbank('NC_012920','SequenceOnly',true)The
getgenbankfunction retrieves the nucleotide sequence from the GenBank database and creates a character array.mitochondria = GATCACAGGTCTATCACCCTATTAACCACTCACGGGAGCTCTCCATGCAT TTGGTATTTTCGTCTGGGGGGTGTGCACGCGATAGCATTGCGAGACGCTG GAGCCGGAGCACCCTATGTCGCAGTATCTGTCTTTGATTCCTGCCTCATT CTATTATTTATCGCACCTACGTTCAATATTACAGGCGAACATACCTACTA AAGT . . .
If you don't have a Web connection, you can load the data from a MAT file included with the Bioinformatics Toolbox™ software, using the command
load mitochondria
The
loadfunction loads the sequencemitochondriainto the MATLAB Workspace.Get information about the sequence. Type
whos mitochondria
Information about the size of the sequence displays in the MATLAB Command Window.
Name Size Bytes Class Attributes mitochondria 1x16569 33138 char
Determining Nucleotide Composition
The following procedure illustrates how to determine the monomers and dimers, and then visualize data in graphs and bar plots. Sections of a DNA sequence with a high percent of A+T nucleotides usually indicate intergenic parts of the sequence, while low A+T and higher G+C nucleotide percentages indicate possible genes. Many times high CG dinucleotide content is located before a gene.
After you read a sequence into the MATLAB environment, you can use the sequence statistics functions to determine if your sequence has the characteristics of a protein-coding region. This procedure uses the human mitochondrial genome as an example. See Reading Sequence Information from the Web.
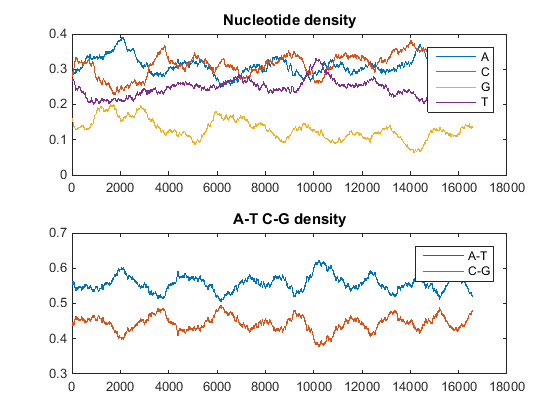
Plot monomer densities and combined monomer densities in a graph. In the MATLAB Command Window, type
ntdensity(mitochondria)
This graph shows that the genome is A+T rich.
Count the nucleotides using the
basecountfunction.basecount(mitochondria)
A list of nucleotide counts is shown for the 5'-3' strand.
ans = A: 5124 C: 5181 G: 2169 T: 4094Count the nucleotides in the reverse complement of a sequence using the
seqrcomplementfunction.basecount(seqrcomplement(mitochondria))
As expected, the nucleotide counts on the reverse complement strand are complementary to the 5'-3' strand.
ans = A: 4094 C: 2169 G: 5181 T: 5124Use the function
basecountwith thechartoption to visualize the nucleotide distribution.figure basecount(mitochondria,'chart','pie');
A pie chart displays in the MATLAB Figure window.
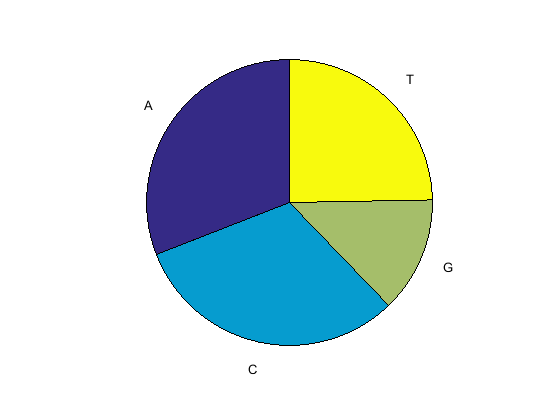
Count the dimers in a sequence and display the information in a bar chart.
figure dimercount(mitochondria,'chart','bar')
ans = AA: 1604 AC: 1495 AG: 795 AT: 1230 CA: 1534 CC: 1771 CG: 435 CT: 1440 GA: 613 GC: 711 GG: 425 GT: 419 TA: 1373 TC: 1204 TG: 513 TT: 1004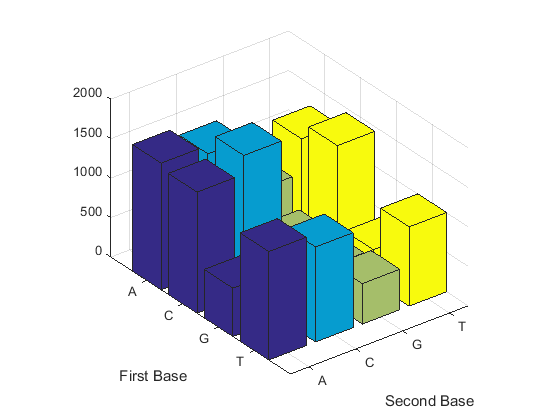
Determining Codon Composition
The following procedure illustrates how to look at codons for the six reading frames. Trinucleotides (codon) code for an amino acid, and there are 64 possible codons in a nucleotide sequence. Knowing the percent of codons in your sequence can be helpful when you are comparing with tables for expected codon usage.
After you read a sequence into the MATLAB environment, you can analyze the sequence for codon composition. This procedure uses the human mitochondria genome as an example. See Reading Sequence Information from the Web.
Count codons in a nucleotide sequence. In the MATLAB Command Window, type
codoncount(mitochondria)
The codon counts for the first reading frame displays.
AAA - 167 AAC - 171 AAG - 71 AAT - 130 ACA - 137 ACC - 191 ACG - 42 ACT - 153 AGA - 59 AGC - 87 AGG - 51 AGT - 54 ATA - 126 ATC - 131 ATG - 55 ATT - 113 CAA - 146 CAC - 145 CAG - 68 CAT - 148 CCA - 141 CCC - 205 CCG - 49 CCT - 173 CGA - 40 CGC - 54 CGG - 29 CGT - 27 CTA - 175 CTC - 142 CTG - 74 CTT - 101 GAA - 67 GAC - 53 GAG - 49 GAT - 35 GCA - 81 GCC - 101 GCG - 16 GCT - 59 GGA - 36 GGC - 47 GGG - 23 GGT - 28 GTA - 43 GTC - 26 GTG - 18 GTT - 41 TAA - 157 TAC - 118 TAG - 94 TAT - 107 TCA - 125 TCC - 116 TCG - 37 TCT - 103 TGA - 64 TGC - 40 TGG - 29 TGT - 26 TTA - 96 TTC - 107 TTG - 47 TTT - 78
Count the codons in all six reading frames and plot the results in heat maps.
for frame = 1:3 figure subplot(2,1,1); codoncount(mitochondria,'frame',frame,'figure',true,... 'geneticcode','Vertebrate Mitochondrial'); title(sprintf('Codons for frame %d',frame)); subplot(2,1,2); codoncount(mitochondria,'reverse',true,'frame',frame,... 'figure',true,'geneticcode','Vertebrate Mitochondrial'); title(sprintf('Codons for reverse frame %d',frame)); endHeat maps display all 64 codons in the 6 reading frames.

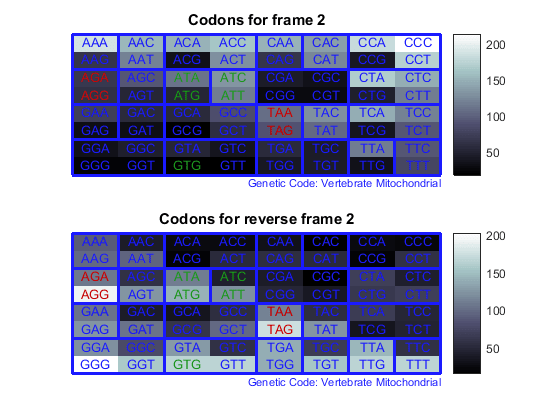
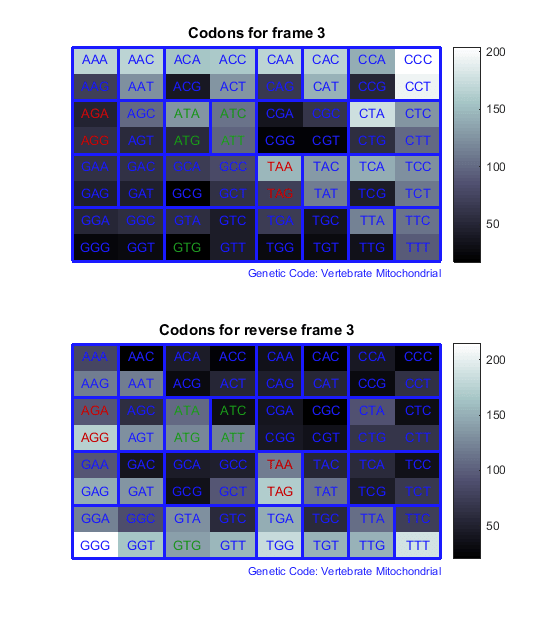
Open Reading Frames
The following procedure illustrates how to locate the open reading frames using a specific genetic code. Determining the protein-coding sequence for a eukaryotic gene can be a difficult task because introns (noncoding sections) are mixed with exons. However, prokaryotic genes generally do not have introns and mRNA sequences have the introns removed. Identifying the start and stop codons for translation determines the protein-coding section, or open reading frame (ORF), in a sequence. Once you know the ORF for a gene or mRNA, you can translate a nucleotide sequence to its corresponding amino acid sequence.
After you read a sequence into the MATLAB environment, you can analyze the sequence for open reading frames. This procedure uses the human mitochondria genome as an example. See Reading Sequence Information from the Web.
Display open reading frames (ORFs) in a nucleotide sequence. In the MATLAB Command Window, type:
seqshoworfs(mitochondria);
If you compare this output to the genes shown on the NCBI page for
NC_012920, there are fewer genes than expected. This is because vertebrate mitochondria use a genetic code slightly different from the standard genetic code. For a list of genetic codes, see the Genetic Code table in theaa2ntreference page.Display ORFs using the
Vertebrate Mitochondrialcode.orfs= seqshoworfs(mitochondria,... 'GeneticCode','Vertebrate Mitochondrial',... 'alternativestart',true);Notice that there are now two large ORFs on the third reading frame. One starts at position 4470 and the other starts at 5904. These correspond to the genes ND2 (NADH dehydrogenase subunit 2 [Homo sapiens] ) and COX1 (cytochrome c oxidase subunit I) genes.
Find the corresponding stop codon. The start and stop positions for ORFs have the same indices as the start positions in the fields
StartandStop.ND2Start = 4470; StartIndex = find(orfs(3).Start == ND2Start) ND2Stop = orfs(3).Stop(StartIndex)
The stop position displays.
ND2Stop = 5511Using the sequence indices for the start and stop of the gene, extract the subsequence from the sequence.
ND2Seq = mitochondria(ND2Start:ND2Stop)
The subsequence (protein-coding region) is stored in
ND2Seqand displayed on the screen.attaatcccctggcccaacccgtcatctactctaccatctttgcaggcac actcatcacagcgctaagctcgcactgattttttacctgagtaggcctag aaataaacatgctagcttttattccagttctaaccaaaaaaataaaccct cgttccacagaagctgccatcaagtatttcctcacgcaagcaaccgcatc cataatccttc . . .
Determine the codon distribution.
codoncount (ND2Seq)
The codon count shows a high amount of
ACC,ATA,CTA, andATC.AAA - 10 AAC - 14 AAG - 2 AAT - 6 ACA - 11 ACC - 24 ACG - 3 ACT - 5 AGA - 0 AGC - 4 AGG - 0 AGT - 1 ATA - 23 ATC - 24 ATG - 1 ATT - 8 CAA - 8 CAC - 3 CAG - 2 CAT - 1 CCA - 4 CCC - 12 CCG - 2 CCT - 5 CGA - 0 CGC - 3 CGG - 0 CGT - 1 CTA - 26 CTC - 18 CTG - 4 CTT - 7 GAA - 5 GAC - 0 GAG - 1 GAT - 0 GCA - 8 GCC - 7 GCG - 1 GCT - 4 GGA - 5 GGC - 7 GGG - 0 GGT - 1 GTA - 3 GTC - 2 GTG - 0 GTT - 3 TAA - 0 TAC - 8 TAG - 0 TAT - 2 TCA - 7 TCC - 11 TCG - 1 TCT - 4 TGA - 10 TGC - 0 TGG - 1 TGT - 0 TTA - 8 TTC - 7 TTG - 1 TTT - 8
Look up the amino acids for codons
ATA,CTA,ACC, andATC.aminolookup('code',nt2aa('ATA')) aminolookup('code',nt2aa('CTA')) aminolookup('code',nt2aa('ACC')) aminolookup('code',nt2aa('ATC'))The following displays:
Ile isoleucine Leu leucine Thr threonine Ile isoleucine
Amino Acid Conversion and Composition
The following procedure illustrates how to extract the protein-coding sequence from a gene sequence and convert it to the amino acid sequence for the protein. Determining the relative amino acid composition of a protein will give you a characteristic profile for the protein. Often, this profile is enough information to identify a protein. Using the amino acid composition, atomic composition, and molecular weight, you can also search public databases for similar proteins.
After you locate an open reading frame (ORF) in a gene, you can convert it to an amino sequence and determine its amino acid composition. This procedure uses the human mitochondria genome as an example. See Open Reading Frames.
Convert a nucleotide sequence to an amino acid sequence. In this example, only the protein-coding sequence between the start and stop codons is converted.
ND2AASeq = nt2aa(ND2Seq,'geneticcode',... 'Vertebrate Mitochondrial')The sequence is converted using the
Vertebrate Mitochondrialgenetic code. Because the propertyAlternativeStartCodonsis set to'true'by default, the first codonattis converted toMinstead ofI.MNPLAQPVIYSTIFAGTLITALSSHWFFTWVGLEMNMLAFIPVLTKKMNP RSTEAAIKYFLTQATASMILLMAILFNNMLSGQWTMTNTTNQYSSLMIMM AMAMKLGMAPFHFWVPEVTQGTPLTSGLLLLTWQKLAPISIMYQISPSLN VSLLLTLSILSIMAGSWGGLNQTQLRKILAYSSITHMGWMMAVLPYNPNM TILNLTIYIILTTTAFLLLNLNSSTTTLLLSRTWNKLTWLTPLIPSTLLS LGGLPPLTGFLPKWAIIEEFTKNNSLIIPTIMATITLLNLYFYLRLIYST SITLLPMSNNVKMKWQFEHTKPTPFLPTLIALTTLLLPISPFMLMIL
Compare your conversion with the published conversion in the GenPept database.
ND2protein = getgenpept('YP_003024027','sequenceonly',true)The
getgenpeptfunction retrieves the published conversion from the NCBI database and reads it into the MATLAB Workspace.Count the amino acids in the protein sequence.
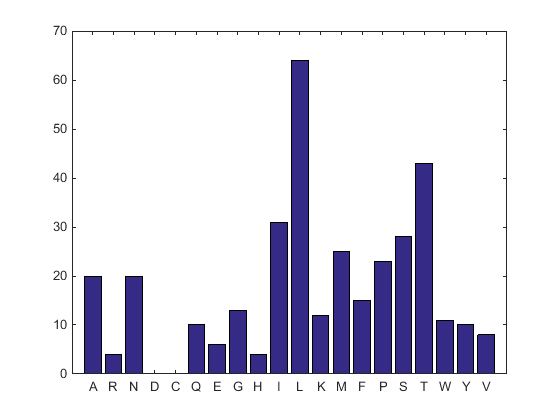
aacount(ND2AASeq, 'chart','bar')
A bar graph displays. Notice the high content for leucine, threonine and isoleucine, and also notice the lack of cysteine and aspartic acid.
Determine the atomic composition and molecular weight of the protein.
atomiccomp(ND2AASeq) molweight (ND2AASeq)
The following displays in the MATLAB Workspace:
ans = C: 1818 H: 2882 N: 420 O: 471 S: 25ans = 3.8960e+004
If this sequence was unknown, you could use this information to identify the protein by comparing it with the atomic composition of other proteins in a database.