metafeatures
Attractor metagene algorithm for feature engineering using mutual information-based learning
Syntax
Description
M = metafeatures(X)M in X using
the attractor metagene algorithm described in [1].
M is a r-by-n matrix. r is
the number of metafeatures identified during each repetition of the
algorithm. The default number of repetitions is 1. By default, only
unique metafeatures are returned in M. If multiple repetitions result
in the same metafeature, then just one copy is returned in M. n is
the number of samples (patients or time points).
X is a p-by-n numeric
matrix. p is the number of variables, features,
or genes. In other words, rows of X correspond
to variables, such as measurements of gene expression for different
genes. Columns correspond to different samples, such as patients or
time points.
[
uses a p-by-1 cell array of character vectors or string vector
M,W,GSorted]
= metafeatures(X,G)G containing the variable names and returns a
p-by-r cell array of variable names
GSorted sorted by the decreasing weight.
The ith column of GSorted lists
the feature (variable) names in order of their contributions to the ith
metafeature.
[ returns the indices M,W,GSorted,GSortedInd]
= metafeatures(___)GSortedInd such
that GSorted = G(GSortedInd).
[___] = metafeatures(___,
uses additional options specified by one or more Name,Value)Name,Value pair
arguments.
[___] = metafeatures( uses
a p-by-n table T)T.
Gene names are the row names of the table. M = W'*T{:,:}.
[___] = metafeatures(
uses additional options specified by one or more T,Name,Value)Name,Value pair
arguments.
Note
It is possible that the number of metafeatures (r)
returned in M can be fewer than the number of
replicates (repetitions). Even though you may have set the number
of replicates to a positive integer greater than 1, if each repetition
returns the same metafeature, then r is 1, and M is
1-by-n. This is because, by default, the function
returns only unique metafeatures. If you prefer to get all metafeatures,
set 'ReturnUnique' to false.
A metafeature is considered unique if the Pearson correlation between
it and all previously found metafeatures is less than the 'UniqueTolerance' value
(the default value is 0.98).
Examples
Load the breast cancer gene expression data. The data
was retrieved from the Cancer Genome Atlas (TCGA) on May 20, 2014
and contains gene expression data of 17814 genes for 590 different
patients. The expression data is stored in the variable geneExpression.
The gene names are stored in the variable geneNames.
load TCGA_Breast_Gene_ExpressionThe data has several NaN values.
sum(sum(isnan(geneExpression)))
ans =
1695Use the k-nearest neighbor imputation method to replace missing data with the corresponding value from an average of the k columns that are nearest.
geneExpression = knnimpute(geneExpression,3);
There are three common drivers of breast cancer:
ERBB2, estrogen, and progestrone.
metafeatures allows you to
seed the starting weights to focus on the genes of
interest. In this case, set the weight for each of
these genes to 1 in three different rows of
startValues. Each row
corresponds to initial values for a different
replicate (repetition).
erbb = find(strcmp('ERBB2',geneNames)); estrogen = find(strcmp('ESR1',geneNames)); progestrone = find(strcmp('PGR',geneNames)); startValues = zeros(size(geneExpression,1),3); startValues(erbb,1) = 1; startValues(estrogen,2) = 1; startValues(progestrone,3) = 1;
Apply the attractor metagene algorithm to the imputed data.
[meta, weights, genes_sorted] = metafeatures(geneExpression,geneNames,'start',startValues);
The variable meta has the value of
three metagenes discovered for each sample. Plot these three metagenes
to gain insight into the nature of gene regulation across different
phenotypes of breast cancer.
plot3(meta(1,:),meta(2,:),meta(3,:),'o') xlabel('ERBB2 metagene') ylabel('Estrogen metagene') zlabel('Progestrone metagene')
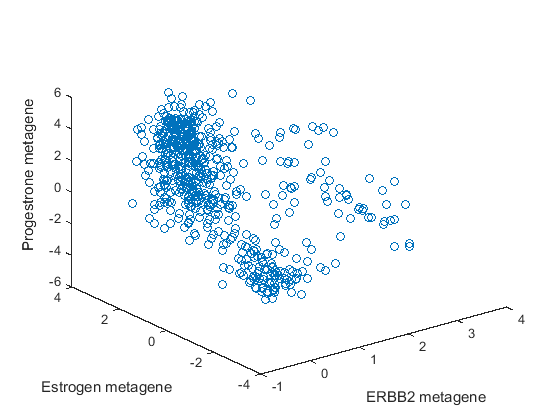
Based on the plot, observe the following.
There is a group of points clustered together with low values for all three metagenes. Based on mRNA levels, these could be triple-negative or basal type breast cancer.
There is a group of points that have high estrogen receptor metagene expression and span across both high and low progestrone metagene expression. There are no points with high progestrone metagene expression and low estrogen metagene expression. This is consistent with the observation that ER-/PR+ breast cancers are extremely rare [3].
The remaining points are the ERBB2 positive cancers. They have less representation in this data set than the hormone-driven and triple negative cancers.
Input Arguments
Name-Value Arguments
Output Arguments
More About
References
[1] Cheng, W-Y., Ou Yang, T-H., and Anastassiou, D. (2013). Biomolecular events in cancer revealed by attractor metagenes. PLoS Computational Biology 9(2): e1002920.
[2] Daub, C., Steuer, R., Selbig, J., and Kloska, S. (2004). Estimating mutual information using B-spline functions – an improved similarity measure for analysing gene expression data. BMC Bioinformatics 5, 118.
[3] Hefti, M.M., Hu, R., Knoblauch, N.W., Collins, L.C., Haibe-Kains, B., Tamimi, R.M., and Beck, A.H. (2013). Estrogen receptor negative/progesterone receptor positive breast cancer is not a reproducible subtype. Breast Cancer Research. 15:R68.